



May 8 is observed as World Thalassemia Day every year. The day commemorates the struggles of the patients suffering from this genetic disorder and raise awareness about the disease and its symptoms. This global observance also honours the longstanding efforts of doctors, other medical staff dedicated to better the lives of patients from the disease and scientists bringing new advancements in the field to eradicate the disease.
In the year 1994, the Thalassemia International Federation established May 8 as International Thalassemia Day. The day was declared an International observance in the loving memory of George Englezos and all thalassemia patients who fought the disease and while confronting the social stigma attached to it achieved both personal and professional success in life. George was the son of thalassemia International Federation’s (TIF) president and founder, Panos Englezos and worked as a scientist.
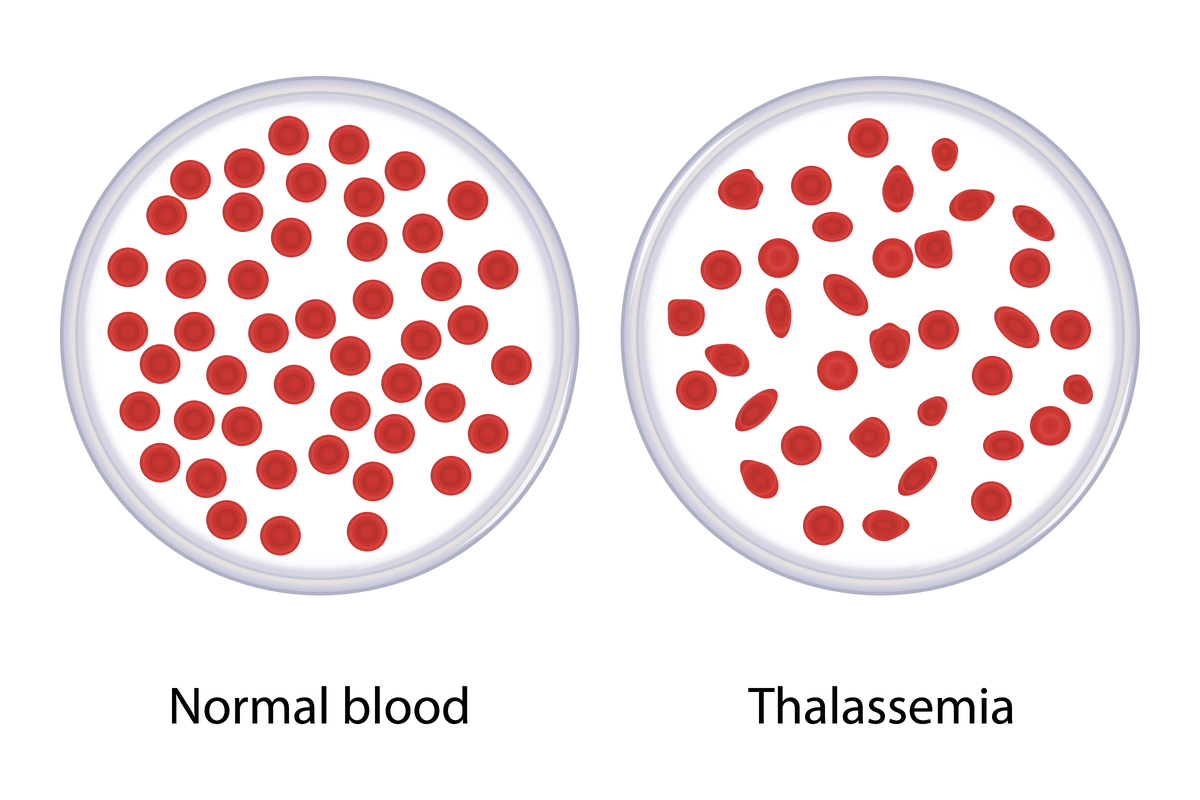
Thalassemia is a genetic blood disorder that passes down the generations from parents to children. The disease significantly reduces the haemoglobin count in the body and affects the production of red blood cells. The severity of the disease depends upon the type Thalassemia.
Symptoms
There are several types of thalassemia. The signs and symptoms you have depend on the type and severity of your condition.
Thalassemia signs and symptoms can include:
- Fatigue
- Weakness
- Pale or yellowish skin
- Facial bone deformities
- Slow growth
- Abdominal swelling
- Dark urine
Some babies show signs and symptoms of thalassemia at birth; others develop them during the first two years of life. Some people who have only one affected hemoglobin gene don’t have thalassemia symptoms.
Causes
Thalassemia is caused by mutations in the DNA of cells that make hemoglobin — the substance in red blood cells that carries oxygen throughout your body. The mutations associated with thalassemia are passed from parents to children.
Hemoglobin molecules are made of chains called alpha and beta chains that can be affected by mutations. In thalassemia, the production of either the alpha or beta chains are reduced, resulting in either alpha-thalassemia or beta-thalassemia.
In alpha-thalassemia, the severity of thalassemia you have depends on the number of gene mutations you inherit from your parents. The more mutated genes, the more severe your thalassemia.
In beta-thalassemia, the severity of thalassemia you have depends on which part of the hemoglobin molecule is affected.
Alpha-thalassemia
Four genes are involved in making the alpha hemoglobin chain. You get two from each of your parents. If you inherit:
- One mutated gene, you’ll have no signs or symptoms of thalassemia. But you are a carrier of the disease and can pass it on to your children.
- Two mutated genes, your thalassemia signs and symptoms will be mild. This condition might be called alpha-thalassemia trait.
- Three mutated genes, your signs and symptoms will be moderate to severe.
Inheriting four mutated genes is rare and usually results in stillbirth. Babies born with this condition often die shortly after birth or require lifelong transfusion therapy. In rare cases, a child born with this condition can be treated with transfusions and a stem cell transplant.
Beta-thalassemia
Two genes are involved in making the beta hemoglobin chain. You get one from each of your parents. If you inherit:
- One mutated gene, you’ll have mild signs and symptoms. This condition is called thalassemia minor or beta-thalassemia.
- Two mutated genes, your signs and symptoms will be moderate to severe. This condition is called thalassemia major, or Cooley anemia.
Babies born with two defective beta hemoglobin genes usually are healthy at birth but develop signs and symptoms within the first two years of life. A milder form, called thalassemia intermedia, also can result from two mutated genes.
Risk factors
Factors that increase your risk of thalassemia include:
- Family history of thalassemia. Thalassemia is passed from parents to children through mutated hemoglobin genes.
- Certain ancestry. Thalassemia occurs most often in African Americans and in people of Mediterranean and Southeast Asian descent.
Complications
Possible complications of moderate to severe thalassemia include:
- Iron overload. People with thalassemia can get too much iron in their bodies, either from the disease or from frequent blood transfusions. Too much iron can result in damage to your heart, liver and endocrine system, which includes hormone-producing glands that regulate processes throughout your body.
- Infection. People with thalassemia have an increased risk of infection. This is especially true if you’ve had your spleen removed.
In cases of severe thalassemia, the following complications can occur:
- Bone deformities. Thalassemia can make your bone marrow expand, which causes your bones to widen. This can result in abnormal bone structure, especially in your face and skull. Bone marrow expansion also makes bones thin and brittle, increasing the chance of broken bones.
- Enlarged spleen. The spleen helps your body fight infection and filter unwanted material, such as old or damaged blood cells. Thalassemia is often accompanied by the destruction of a large number of red blood cells. This causes your spleen to enlarge and work harder than normal.
An enlarged spleen can make anemia worse, and it can reduce the life of transfused red blood cells. If your spleen grows too big, your doctor might suggest surgery to remove it.
- Slowed growth rates. Anemia can both slow a child’s growth and delay puberty.
- Heart problems. Congestive heart failure and abnormal heart rhythms can be associated with severe thalassemia.
Prevention
In most cases, you can’t prevent thalassemia. If you have thalassemia, or if you carry a thalassemia gene, consider talking with a genetic counselor for guidance if you want to have children.
There is a form of assisted reproductive technology diagnosis, which screens an embryo in its early stages for genetic mutations combined with in vitro fertilization. This might help parents who have thalassemia or who are carriers of a defective hemoglobin gene have healthy babies.
The procedure involves retrieving mature eggs and fertilizing them with sperm in a dish in a laboratory. The embryos are tested for the defective genes, and only those without genetic defects are implanted into the uterus.